Rückstau strapaziert die Haut
Stauungsdermatitis ganzheitlich angehen
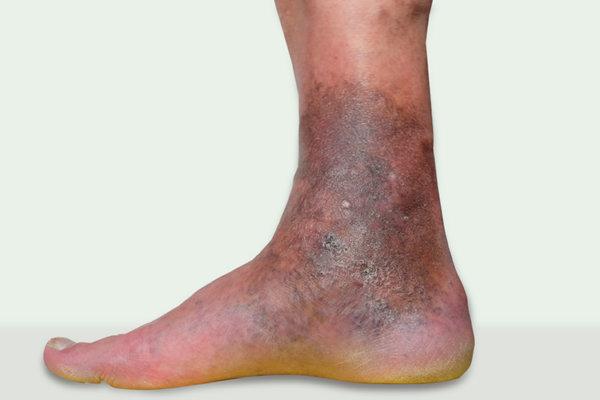
Bei Schmerzen, Juckreiz und Entzündungen an den Unterschenkeln kann es sich um eine eine Stauungsdermatitis im Zusammenhang mit chronischer venöser Insuffizienz handeln. Die Diagnose ist jedoch nicht immer eindeutig, und die Behandlung erfordert ein ganzheitliches Vorgehen, wie eine aktuelle Übersichtsarbeit in BMJ zusammenfasst.